


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Gu, B.; 高橋 三郎*; 前川 禎通
Physical Review B, 96(21), p.214423_1 - 214423_6, 2017/12
被引用回数:9 パーセンタイル:42.75(Materials Science, Multidisciplinary)Using density functional theory calculations, we have found an enhanced magneto-optical Kerr effect in Fe/insulator interfaces. The results of our study indicate that interfacial Fe atoms in the Fe films have a lowdimensional nature, which causes the following two effects: (1) The diagonal component 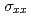 of the optical conductivity decreases dramatically because the hopping integral for electrons between Fe atoms is suppressed by the low dimensionality. (2) The off-diagonal component
of the optical conductivity decreases dramatically because the hopping integral for electrons between Fe atoms is suppressed by the low dimensionality. (2) The off-diagonal component 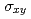 of the optical conductivity does not change at low photon energies, but it is enhanced at photon energies around 2 eV, where we obtain enhanced orbital magnetic moments and spin-orbit correlations for the interfacial Fe atoms. A large Kerr angle develops in proportion to the ratio /. Our findings indicate an efficient way to enhance the effect of spin-orbit coupling at metal/insulator interfaces without using heavy elements.
of the optical conductivity does not change at low photon energies, but it is enhanced at photon energies around 2 eV, where we obtain enhanced orbital magnetic moments and spin-orbit correlations for the interfacial Fe atoms. A large Kerr angle develops in proportion to the ratio /. Our findings indicate an efficient way to enhance the effect of spin-orbit coupling at metal/insulator interfaces without using heavy elements.
Herv du Penhoat, M.-A.*; Kamol Ghose, K.*; Gaigeot, M.-P.*; Vuilleumier, R.*; 藤井 健太郎; 横谷 明徳; Politis, M.-F.*
du Penhoat, M.-A.*; Kamol Ghose, K.*; Gaigeot, M.-P.*; Vuilleumier, R.*; 藤井 健太郎; 横谷 明徳; Politis, M.-F.*
Physical Chemistry Chemical Physics, 17(48), p.32375 - 32383, 2015/12
被引用回数:8 パーセンタイル:31.65(Chemistry, Physical)We have investigated the gas phase fragmentation dynamics following the core ionisation of 2-deoxy-D-ribose (dR), a major component in the DNA chain. To that aim, we use state-of-the-art ab initio Density Functional Theory-based Molecular Dynamics simulations. The ultrafast dissociation dynamics of the core-ionised biomolecule, prior Auger decay, is first modelled for 10 fs to generate initial configurations (atomic positions and velocities) for the subsequent dynamics of the doubly ionised biomolecule in the ground state. The furanose, linear and pyranose conformations of dR were investigated. We show that fragmentation is relatively independent of the atom struck or of the duration of the core vacancy, but depends rather critically on the molecular orbital removed following Auger decay.
千原 順三*; 山極 満
Progress of Theoretical Physics, 111(3), p.339 - 359, 2004/03
被引用回数:5 パーセンタイル:37.3(Physics, Multidisciplinary)密度汎関数理論は外部ポテンシャルの下での相互作用系の特性を、対応する非相互作用系と関連付けることにより計算する簡便な手法を提供する。ここでは、この非相互作用系の幾つかの関係式を見いだし、中性の電子-原子核混合系に対する熱力学関係式を、非相互作用系の諸量及び交換相関効果を用いて記述する。これにより、原子核に及ぼされる力の定理が容易に証明される。
 and its homologues
and its homologuesAnton, J.*; 平田 勝; Fricke, B.*; Pershina, V.*
Chemical Physics Letters, 380(1-2), p.95 - 98, 2003/10
被引用回数:6 パーセンタイル:18.14(Chemistry, Physical)われわれはスピン分極を考慮した相対論密度汎関数法の開発を行い、同法を用いてラザホージウム及び同族元素の4塩化物の電子状態を調べた。スピン分極を考慮することにより、同族元素の4塩化物の構造を良好に再現することができたほか、実験的にまだ調べられていないラザホージウム4塩化物の構造予測を行った。
 CHO
CHO CH+HCO; Direct ab initio molecular dynamics study
CH+HCO; Direct ab initio molecular dynamics study黒崎 譲; 横山 啓一
Chemical Physics Letters, 371(5-6), p.568 - 575, 2003/04
被引用回数:31 パーセンタイル:69.48(Chemistry, Physical)UB3LYP/cc-pVDZレベルでの直接非経験的分子動力学法を用いて、T ポテンシャル面上での光分解反応,CHCHOCH+HCO、について全部で400本のトラジェクトリを計算した。その結果、反応生成物であるCHは振動,回転ともに励起しないが、HCOは振動励起しないものの回転励起することが予測された。トラジェクトリ計算の結果を平均するとHCOの回転エネルギーは1.1kcal/molとなり、これは利用可能なエネルギー、7.3kcal/molの15.1%にあたる。本計算結果は実測値と数%の誤差で一致している。
ポテンシャル面上での光分解反応,CHCHOCH+HCO、について全部で400本のトラジェクトリを計算した。その結果、反応生成物であるCHは振動,回転ともに励起しないが、HCOは振動励起しないものの回転励起することが予測された。トラジェクトリ計算の結果を平均するとHCOの回転エネルギーは1.1kcal/molとなり、これは利用可能なエネルギー、7.3kcal/molの15.1%にあたる。本計算結果は実測値と数%の誤差で一致している。
and homologues in the framework of relativistic density functionaltheoryVarga, S.*; Fricke, B.*; 平田 勝; Bastug, T.; Pershina, V.*; Frizsche, S.*
Journal of Chemical Physics, 104(27), p.6495 - 6498, 2000/06
相対論密度汎関数を用いてRf,Zr,Hf及びTiの4塩化物の全エネルギー計算を行った。金属原子と塩素原子間の距離は、RLDA計算による推定値と実験結果が良好に一致した。また、全エネルギー計算では、実験値と比較して、やや大きいエネルギー値をとるものの、実験的に得られている結合エネルギーの傾向を再現することができた。これらの結果から、相対論密度汎関数法による超アクチノイド元素の化学的性質予測研究の有用性を示すことができた。
Erkoc, S.*; Bastug, T.*; 平田 勝; 館盛 勝一
Chemical Physics Letters, 321(3-4), p.321 - 327, 2000/04
被引用回数:0 パーセンタイル:0.01(Chemistry, Physical)密度汎関数法を用いて表面反応を解析するためには、クラスターサイズと電子状態の関連を明らかにしておく必要がある。本研究では、リチウム金属(001)表面に酸素原子を配置し、酸素上の有効電荷及び酸素原子の結合エネルギーのサイズ依存性を調べた。その結果、リチウム原子20個で構成されるクラスター以上では、酸素原子の有効電荷のサイズ依存性がなくなることが明らかになり、表面反応解析のためのクラスターモデルとして最適であることがわかった。
Bastug, T.*; Erkoc, S.*; 平田 勝; 館盛 勝一
Physical Review A, 95(5), p.3690 - 3694, 1999/05
一連のランタノイド元素の中で、ルテチウム(Lu)は4f軌道が閉殻であり、その凝集挙動を調べることはクラスター科学にとって大変興味深い。われわれは、まず初めに相対論密度汎関数法(RDFT)を用いて、Lu2原子分子の構造最適化計算を行い、ポテンシャエネルギー曲線を作成した。その結果、Laと比較して結合エネルギーは弱く、平衡核間距離もより短いところで安定になることがわかった。また、このポテンシャルを用いてLu3個から147個のクラスターの分子動力学(MD)シミュレーションを行い、Lu金属クラスターの安定構造を予測した。
Erkoc, S.*; Bastug, T.*; 平田 勝; 館盛 勝一
Journal of the Physical Society of Japan, 68(2), p.440 - 445, 1999/02
相対論密度汎関数法(RDFT)を用いて、ウラン(U)2原子分子の構造最適化計算を行い、ポテンシャルエネルギー曲線を作成した。このポテンシャルを用いてU3個から137個のクラスターの分子動力学(MD)シミュレーションを行った。その結果、U13個で構成されるクラスターが最も対称性が良く(Ih対称)安定に存在する可能性の高いことがわかった。また、結合エネルギーのクラスターサイズ依存性を調べたところ、U3個から13個までのクラスターでは、クラスターサイズの増加とともに急激に結合エネルギーが減少し、それ以降のクラスターサイズではゆるやかに結合エネルギーが減少することがわかった。
神林 奨; 千原 順三
Molecular Simulation, 16, p.31 - 46, 1996/00
被引用回数:4 パーセンタイル:17.31(Chemistry, Physical)従来のカーパリネロの理論による分子動力学法とは異なった、新しい第1原理的分子動力学法(QHNC-MD法)を考案した。QHNC-MD法では、液体金属中の電子及びイオンに関する動径分布関数と有効イオン間ポテンシャルに対する量子的HNC方程式を、古典的分子動力学シミュレーションを用いて解く方法である。この方法では、分子動力学シミュレーションによって得られるイオン間分布関数と有効イオン間ポテンシャルを自己無撞着に決定することが可能である。また、QHNC-MD法による有効イオン間ポテンシャルの収束計算は高速であり、しかも、数千から数万個の規模のシミュレーションが可能である。この点はカーパリネロ手法と大きく異なる部分である。液体アルカリ金属に関するQHNC-MDシミュレーションから得られた静的構造因子は、X線・中性子線実験の結果と極めて良く一致し、従来のQHNC方程式の近似解に見られる欠点を取り除くことが可能となった。
千原 順三; 石飛 昌光*
Molecular Simulation, 12(3-6), p.187 - 195, 1994/00
被引用回数:3 パーセンタイル:14.84(Chemistry, Physical)イオン電子混合系とみなせる液体金属においては、その分子動力学を行うとき、常に2体の原子間ポテンシャルで正確に記述できることを示した(多体力は不要)。
大和田 謙; 鈴木 和弥
Spectrochimica Acta, Part A, 50(6), p.1057 - 1063, 1994/00
赤外・ラマンスペクトルデータの基準振動解析から得られる二次のポテンシャル定数(力の定数)を応用して、多原子分子形成時の電子化学ポテンシャルを求める方法を検討した。本報では密度汎関数理論を採用して、これに単純結合一電荷モデル(Simple Bond-Charge Model)を試験的に組み込んだ近似法を提案した。この方法を用いて、種々の異核二原子分子及び多原子分子の電子化学ポテンシャルを計算し、実験値及びab-initio SCF計算から得られる値と比較した結果、本法の有用性を確かめることができた。
大和田 謙
Journal of Physical Chemistry, 97(9), p.1832 - 1834, 1993/00
被引用回数:10 パーセンタイル:41.34(Chemistry, Physical)密度汎関数理論において、分子中の各原子の電子エネルギーが電子数と核ポテンシャルについて汎関数展開できることを利用し、多原子分子系の電子化学ポテンシャルを評価するための基本式を導いた。これを用いて種々の多原子分子の電子化学ポテンシャルを計算し、実験値及びサンダーソンの原理から得られる値と比較した結果、基本式の有用性を確かめることができた。
大和田 謙
Spectrochimica Acta, Part A, 49(1), p.81 - 94, 1993/00
異核=原子分子の基準振動解析から得られる調和および非調和ポテンシャル定数を応用して、分子の化学ポテンシャルを求める方法を検討した。本法は主として密度汎関数理論に基礎を置き、分子中の各原子のエネルギーが電子数と核ポテンシャルに関して展開できることを利用する。これらの展開は、N個の電子数と与えられた核ポテンシャルv(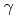 )をもつ系の基底状態の全エネルギーがN,v()の汎関数として表されるので可能となる。本法を用いて、多数のアルカリハライド分子、その他の異核=原子分子の化学ポテンシャルを計算し、Sandersonの原理及びab-initio SCF計算から得られる値と比較した結果、本法の有用性を確かめることができた。また見掛けの化学ハードニス及び福井関数を含む積分項の値を評価した。分子形成時の化学ポテンシャル変化についても簡単に議論した。
)をもつ系の基底状態の全エネルギーがN,v()の汎関数として表されるので可能となる。本法を用いて、多数のアルカリハライド分子、その他の異核=原子分子の化学ポテンシャルを計算し、Sandersonの原理及びab-initio SCF計算から得られる値と比較した結果、本法の有用性を確かめることができた。また見掛けの化学ハードニス及び福井関数を含む積分項の値を評価した。分子形成時の化学ポテンシャル変化についても簡単に議論した。
大和田 謙
Journal of Physical Chemistry, 96(14), p.5825 - 5829, 1992/00
被引用回数:2 パーセンタイル:11.73(Chemistry, Physical)異核二原子分子の基準振動解析から得られる調和および非調和ポテンシャル定数を応用して、分子形成時の各原子の化学ポテンシャル変化と分子の化学ポテンシャルを求める方法を検討した。本法は主として密度汎関数理論に基礎を置き、分子中の各原子のエネルギーが電子数と核ポテンシャルに関して展開できることを利用する。これらの展開は、N個の電子数と与えられた核ポテンシャルv(F)をもつ系の基底状態の全エネルギーがN,v(F)の汎関数として表されるので可能となる。本法を用いて、多数のアルカリハライド分子、その他の異核二原子分子の原子および分子の化学ポテンシャルを計算し、Sandersonの原理から得られる値と比較した結果、本法の有用性を確かめることができた。
大和田 謙
Journal of Chemical Physics, 78(3), p.1414 - 1419, 1983/00
被引用回数:6 パーセンタイル:28.89(Chemistry, Physical)密度汎関数理論における電子運動エネルギー汎関数を見い出すために、まず原子に対する一体の密度行列を考察し、これから指数関数的に減少する近似関数形を提案した。ここでnは主量子数を表わし、P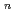 は一体の密度行列で次の式によって与えられる。(2)式でAn,Bn,Cnはそれぞれと'の関数(slowly varying functions)であり、fnは主量子数に依存する関数である。(1)および(2)式を用いて目的の電子運動エネルギー汎関数が導かれ、この式は電子密度の核カスプ挙動および漸近挙動を記述するのに妥当なものであり、また原子の電子運動エネルギーの計算に役立つことが示された。
は一体の密度行列で次の式によって与えられる。(2)式でAn,Bn,Cnはそれぞれと'の関数(slowly varying functions)であり、fnは主量子数に依存する関数である。(1)および(2)式を用いて目的の電子運動エネルギー汎関数が導かれ、この式は電子密度の核カスプ挙動および漸近挙動を記述するのに妥当なものであり、また原子の電子運動エネルギーの計算に役立つことが示された。
佐久間 博*; 舘 幸男; 四辻 健治; 河村 雄行*
no journal, ,
粘土鉱物は、大きな比表面積と高い吸着能を持つため、自然環境中の有害物質に対する良い吸着材と成りうる。イオンや分子の、層間および外部表面における吸着サイトは、分析試験や比較的簡単なモデルを用いた分子シミュレーションにより評価できる。一方、エッジ表面での吸着サイトについては、エッジ構造のモデル化がまだ確立していないため、十分な理解が得られていない。そこで本研究では、モンモリロナイトのエッジ構造について、密度汎関数理論に基づく第一原理計算手法で調べた。表面エネルギーを計算することにより、4種類のエッジ表面に対し、同形置換、層電荷、及び層間陽イオンの位置の影響について評価した。さらに、エッジの酸解離定数を計算し、陽イオンの吸着サイトに成り得るかどうかについて議論した。
Gu, B.
no journal, ,
Magneto-optical Kerr effect (MOKE) is the phenomenon in which the light reflected from a magnetized material has a rotated plane of polarization. The MOKE originates from spin-orbit coupling in materials, and the effect has played a significant role in the rapidly developing field of spintronics. For applications to magneto-optical devices, a large MOKE is required. By density functional theory calculations, we have found an enhanced MOKE at Fe/insulator interfaces. Our study indicates that interfacial Fe atoms in the Fe films have a low-dimensional nature, which causes the following two effects: (1) The diagonal component of the optical conductivity decreases dramatically because the hopping integral for electrons between Fe atoms is suppressed by the low dimensionality. (2) The off-diagonal component of the optical conductivity does not change at low photon energies, but it is enhanced at photon energies around 2 eV, where we obtain enhanced orbital magnetic moments and spin-orbit correlations for the interfacial Fe atoms. Our findings indicate an efficient way to enhance the effect of spin-orbit coupling at metal/insulator interfaces without using heavy elements.

